


| 中文名称 |
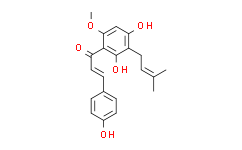
Xanthohumol
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 中文别名 |
呋喃酮丁酸酯;黄腐醇;黄腐醇标准品;黄腐酚;黄腐酚 啤酒花提取物;黄腐酚对照品;啤酒花提取物;(E)-1-[2,4-二羟基-6-甲氧基-3-(3-甲基-2-丁烯基)苯基]-3-(4-羟苯基)丙烯酮;2',4,4'-三羟基-6'-甲氧基-3'-异戊二烯基查尔酮;(E)-1-[2,4-二羟基-6-甲氧基-3-(3-甲基-2-丁烯基)苯基]-3-(4-羟基苯基)丙烯酮;2′,4,4′-三羟基-6′-甲氧基-3′-异戊烯基查耳酮
|
||||||||||||
| 英文名称 |
Xanthohumol
|
||||||||||||
| 英文别名 |
1-(2,4-Dihydroxy-6-methoxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one;Xanthohumo;(E)-1-[2,4-dihydroxy-6-methoxy-3-(3-methylbut-2-enyl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one;2-Propen-1-one,1-[2,4-dihydroxy-6-methoxy-3-(3-methyl-2-buten-1-yl)phenyl]-3-(4-hydroxyphenyl)-,(2E)-;2-Propen-1-one,1-[2,4-dihydroxy-6-methoxy-3-(3-methyl-2-buten-1-yl)phenyl]-3-(4-hydroxyphenyl)...;XANTHOHUMOL(RG);Xanthohumol;XANTHOHUMOL(P);(E)-1-[2,4-Dihydroxy-6-methoxy-3-(3-methyl-2-butenyl)phenyl]-3-(4-hydroxyphenyl)propenone;2',4,4'-Trihydroxy-6'-methoxy-3'-prenylchalcone;(2E)-1-[2,4-Dihydroxy-6-methoxy-3-(3-methyl-2-buten-1-yl)phenyl]-3-(4-hydroxyphenyl)-2-propen-1-one;[
"2'",
"4",
"4'-Trihydroxy-6'-methoxy-3'-prenylchalcone"
];2';T4467YT1NT;1-(2,4-Dihydroxy-6-methoxy-3-(3-methyl-2-butenyl)phenyl)-3-(4-hydroxyphenyl)-2-propen-1-one;(2E)-1-[2,4-dihydroxy-6-methoxy-3-(3-methylbut-2-en-1-yl)phenyl]-3-(4-hydroxyphenyl)prop-2-en-1-one;(E)-1
|
||||||||||||
| Cas No. |
6754-58-1
|
||||||||||||
| 分子式 |
C21H22O5
|
||||||||||||
| 分子量 |
354.40
|
||||||||||||
| 包装储存 |
|
| 生物活性 |
Xanthohumol is one of the principal flavonoids isolated from hops, the inhibitor of diacylglycerol acetyltransferase (DGAT), COX-1 and COX-2, and shows anti-cancer and anti-angiogenic activities. Xanthohumol also has antiviral activity against bovine viral diarrhea virus (BVDV), rhinovirus, HSV-1, HSV-2 and cytomegalovirus (CMV). |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 性状 |
Solid |
||||||||||||
| IC50 & Target[1][2] |
|
||||||||||||
| 体外研究(In Vitro) |
Xanthohumol significantly attenuates ADP-induced blood platelet aggregation, and significantly reduces the expression of fibrinogen receptor (activated form of GPIIbIIIa) on platelets surface. Medlife has not independently confirmed the accuracy of these methods. They are for reference only. |
||||||||||||
| 运输条件 |
Room temperature or refrigerated transportation. |
||||||||||||
| 储存方式 |
|
||||||||||||
| ClinicalTrial |
|
||||||||||||
| 结构分类 | |||||||||||||
| 来源 | |||||||||||||
| 参考文献 |
|
| 溶解度数据 |
体外研究:
DMSO : 83.33 mg/mL (235.13 mM; Need ultrasonic) 配制储备溶液
*
产品不同,其溶解度不同。建议根据产品选择合适的溶剂配制储备溶液;配成溶液后,建议分装保存,避免反复冻融造成的产品失效。 体内研究:
建议根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都建议先按照 体外研究 方式配制澄清的储备液,再依次添加助溶剂:
——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
*
|
|---|
1:一般建议:溶解度为Medlife测试数据,可能与文献描述存在差异。这是由于生产工艺和批次不同产生的正常现象。为了使其更好的溶解,请用37℃加热试管并在超声波水浴中震动片刻。不同批次产品溶解度各有差异,仅做参考,具体以实验方案为准。
2:储存条件:粉末-20°C一般情况可以保存3年,溶于溶剂-80°C一般情况可以保存1年。不同产品及不同批次产品可能存在差异,请细致阅读产品信息,并辅助参考相关文献描述。
Copyright © 2025 陌孚医药 All rights reserved 未经授权禁止拷贝本站所有资料,如有违反,将追究法律责任。
 沪ICP备2023012080号 |
沪ICP备2023012080号 | ![]() 沪公网安备31011402010657号
沪公网安备31011402010657号
 扫码关注公众号
扫码关注公众号
 产品使用指南
产品使用指南
